Thiamine (Vitamin B1)
The gatekeeper between the food you eat and the energy your cells can actually use — and why the form you take determines whether it reaches your brain.
Background: What your cells do with food
Before the thiamine mechanisms make sense, you need a one-paragraph model of how your cells turn food into usable energy.
Every cell runs on ATP — the universal energy currency. To make ATP, cells need to run a sequence of metabolic reactions that happen in two places: first in the cytoplasm (the fluid inside the cell), then inside the mitochondria (the cell's power plants).
Here's the flow for carbohydrates:
flowchart TD
GLU[Glucose] -->|"glycolysis (cytoplasm)<br/>no thiamine needed"| PYR[Pyruvate]
PYR -->|"PDH ★ TPP REQUIRED ★"| ACA[Acetyl-CoA]
ACA -->|"Krebs cycle<br/>α-KGDH ★ TPP REQUIRED ★"| EC["Electron carriers<br/>NADH, FADH₂"]
EC -->|electron transport chain| ATP[ATP + heat + water]
PYR -.->|"if TPP is short,<br/>pyruvate is shunted to"| LAC["Lactate<br/>(fuel that can't be burned)"]
style PYR fill:#5b7Glycolysis runs without thiamine — but the two gates into and through the mitochondria (PDH and α-KGDH) both require the active form, TPP. Starve those gates and pyruvate spills into lactate.
The critical insight: glycolysis doesn't need thiamine, but everything after it does. If you eat carbs and don't have enough thiamine, you produce pyruvate fine — then hit a wall. The pyruvate can't enter the mitochondria efficiently, so it gets shunted sideways into lactate (the waste product of anaerobic metabolism). Your cells are generating fuel that can't be burned properly.
Fats follow a slightly different path (β-oxidation → Acetyl-CoA, which still enters the Krebs cycle) but require far less thiamine per unit of ATP produced. This is why the consequence of thiamine deficiency is felt much more acutely on high-carb diets.
What thiamine actually does
Thiamine in food or supplements is inactive. Before your cells can use it, it must be phosphorylated into its active form: thiamine pyrophosphate (TPP), also called thiamine diphosphate (TDP). This happens inside cells, particularly in the liver and brain.
TPP is a cofactor — it's not the enzyme itself, but without it the enzyme can't work. Three critical enzyme complexes depend on it:
1. Pyruvate Dehydrogenase Complex (PDH)
The gate into the mitochondria for carbohydrate energy.
PDH converts pyruvate → Acetyl-CoA. This is the irreversible step that commits glucose-derived carbon to energy production. If PDH is TPP-limited (due to thiamine deficiency), pyruvate accumulates. It gets converted to lactate by lactate dehydrogenase — the same reaction that causes muscle burn during intense exercise, now happening chronically at rest.
Important nuance: the PDH enzyme protein is preserved even in thiamine deficiency (PMID 3139833) — the enzyme doesn't degrade, it just can't function without its cofactor. This means the limitation is rapidly reversible once thiamine (and specifically TPP) is restored. It also means measuring enzyme protein levels is not a useful marker of thiamine adequacy; you need to measure TPP directly or use the functional transketolase assay.
High-carb meal → high pyruvate load → high PDH demand → high thiamine requirement. This is why populations eating polished white rice (bran removed = B1 stripped) developed beriberi. The bran has the thiamine; the white endosperm has the carbohydrates.
Your personal observation — "makes the most of energy I eat, especially high carb, low fat" — is a direct readout of PDH function. More thiamine → PDH less rate-limited → more pyruvate entering mitochondria → more ATP produced per gram of carbohydrate.
2. Alpha-Ketoglutarate Dehydrogenase (α-KGDH)
The rate-limiting step inside the Krebs cycle — and a neurotransmitter regulator.
α-KGDH converts alpha-ketoglutarate → Succinyl-CoA inside the Krebs cycle. It's not just an energy enzyme. It sits at a metabolic crossroads:
- Glutamate → Alpha-ketoglutarate → Succinyl-CoA — so α-KGDH is part of the pathway that clears glutamate
- Glutamate is the precursor to GABA (the main inhibitory neurotransmitter)
- Glutamate is also directly excitatory when in excess
If α-KGDH is underactive, glutamate builds up. In neurons, excess glutamate causes excitotoxicity — over-activation of NMDA receptors, calcium influx, mitochondrial damage. This is the mechanism behind the neurological damage in Wernicke's encephalopathy (acute thiamine deficiency).
But even sub-clinical insufficiency — not enough to cause Wernicke's, but enough to slow α-KGDH — can shift the glutamate/GABA balance toward excess excitation. The speech and cognitive effects you notice likely have this as a partial mechanism.
α-KGDH is also needed for acetylcholine synthesis: Acetyl-CoA (produced via PDH and the Krebs cycle) is the direct substrate for acetylcholine. Motor neurons, including the brainstem nuclei controlling speech (cranial nerves V, VII, X, XII), are acetylcholine-dependent.
Critically, α-KGDH is more sensitive to thiamine deficiency in brain than PDH (PMID 6149477, PMID 17482317). In thiamine-deficient rats, potassium-stimulated acetylcholine synthesis fell ~75% and KGDHC activity fell 52% in vulnerable thalamic regions — while PDH protein and activity were comparatively preserved. This means the CNS effects of sub-clinical thiamine insufficiency are driven more by α-KGDH than by PDH impairment.
3. Transketolase
The pentose phosphate pathway — cell repair and NADPH production.
Transketolase runs the pentose phosphate pathway (PPP), a side-branch of glycolysis that: - Produces ribose-5-phosphate (needed for DNA/RNA synthesis and repair) - Produces NADPH (needed for glutathione regeneration, the main cellular antioxidant) - Detoxifies reactive oxygen species
The RBC transketolase activation assay (measuring how much enzyme activity improves when you add TPP in the lab) is actually the most sensitive functional test of thiamine status — more sensitive than blood thiamine levels.
Why your brain is especially vulnerable
The brain is metabolically expensive — 2% of body mass, 20% of energy consumption — and runs almost entirely on glucose. This makes it the first organ to notice inadequate PDH or α-KGDH function.
More specifically, thiamine deficiency has predilection for: - Mammillary bodies (memory, Wernicke's signature lesion) - Cerebellum (coordination, speech articulation, timing) - Thalamus (sensory gating, attention) - Brainstem (cranial nerve nuclei for eye movement and speech)
Your speech observation maps directly onto cerebellar and brainstem sensitivity. The cerebellum runs on a precise timing circuit — it coordinates the sequencing of muscle contractions for articulate speech. That circuit is energy-intensive and preferentially damaged by thiamine insufficiency, and preferentially improved when thiamine is adequate.
This also explains why neurological thiamine effects can be noticeable even when you're nowhere near clinical deficiency. Thiamine status exists on a spectrum, and you can be "sub-optimal but not deficient."
Why the supermarket form barely works
Standard thiamine HCl (hydrochloride) and thiamine mononitrate (the form used in fortified foods and cheap supplements) are water-soluble. They are absorbed via dedicated transporters: THTR1 (SLC19A2) and THTR2 (SLC19A3).
The problem is efficiency, not a hard ceiling. THTR2 has extremely high affinity (Km ~25 nM) but low maximum throughput. At physiological dietary levels (1–2 mg/day), active transport handles everything efficiently. As you scale up, a second mechanism — passive diffusion — kicks in and continues absorbing, but at much lower efficiency per mg. A pharmacokinetic study (PMID 22305197) dosing healthy volunteers at 100, 500, and 1500 mg thiamine HCl found plasma levels continued rising at all doses — but the incremental return per mg fell sharply above the active-transport range (~1.5 µM luminal concentration, roughly equivalent to low single-digit mg doses).
This is why anecdotal reports from people taking 100–500 mg thiamine HCl get minimal neurological effect despite the dose: some thiamine is absorbed, but the CNS sees only what can cross the BBB via active transport — a bottleneck on top of a bottleneck. The dose-response curve does not flatten to zero, but the useful CNS fraction of a 500 mg HCl dose is a small fraction of what 25 mg TTFD delivers.
Thiamine mononitrate is the same compound, just a different salt for stability in fortified foods. The bioavailability difference is negligible.
The three forms worth understanding
Benfotiamine — fat-soluble, peripheral specialist
Benfotiamine (S-benzoylthiamine O-monophosphate) is a synthetic thiamine derivative developed in Japan in the 1950s. The key difference: it's fat-soluble, not water-soluble.
Fat-solubility means it can be absorbed via a different route — it passes through intestinal cell membranes directly, rather than depending on the saturating THTR transporters. Plasma thiamine AUC after benfotiamine is approximately 11× higher than equivalent-dose thiamine HCl (PMID 24399744); peak plasma concentration is ~5× higher (PMID 8929745, Loew 1996). The difference between those two figures reflects how benfotiamine extends the absorption curve, not just the peak.
But here's the nuance: benfotiamine is converted to thiamine before it crosses the blood-brain barrier. The phosphatase enzymes in intestinal cells and the liver cleave it to S-benzoylthiamine → thiamine → then it enters blood as regular thiamine. By the time it reaches the brain, it's using the same transporters as thiamine HCl.
So benfotiamine is excellent for: - Peripheral neuropathy (gets high thiamine levels into peripheral nerve tissue) - Anti-glycation — independently from TPP synthesis, benfotiamine blocks three of the four pathways activated by excess glucose: the polyol pathway, the hexosamine pathway, and the PKC pathway. This is relevant for metabolic syndrome and diabetic complications. - Peripheral tissues generally — muscle, nerve, gut
It is not the best choice for primarily neurological/cognitive goals.
Typical dose: 150–300 mg/day for metabolic/peripheral effects.
TTFD (Thiamine Tetrahydrofurfuryl Disulfide) — the brain form
TTFD (thiamine tetrahydrofurfuryl disulfide) is a different class of thiamine derivative: an open-chain disulfide. It's sold as Thiamax (Objective Nutrients, formulated by Elliot Overton).
The chemistry is the key. TTFD has a disulfide bond (–S–S–) attached to the thiamine molecule. This does two things:
-
Makes it fat-soluble and membrane-permeable. Unlike benfotiamine, TTFD doesn't need phosphatase enzymes to "unlock" it at the gut wall. The whole molecule can diffuse passively through cell membranes, including the blood-brain barrier.
-
Once inside cells, it self-converts. Intracellular reducing agents — primarily glutathione and NADH — cleave the disulfide bond. This releases free thiamine directly inside the cell, which is then phosphorylated to TPP. The conversion happens inside neurons, not in the gut or blood.
The practical consequence: TTFD delivers thiamine directly into brain cells without being rate-limited by intestinal transporters or blocked at the BBB. This is why clinical effects — including your speech and cognitive observations — are far more prominent with TTFD than with either thiamine HCl or benfotiamine.
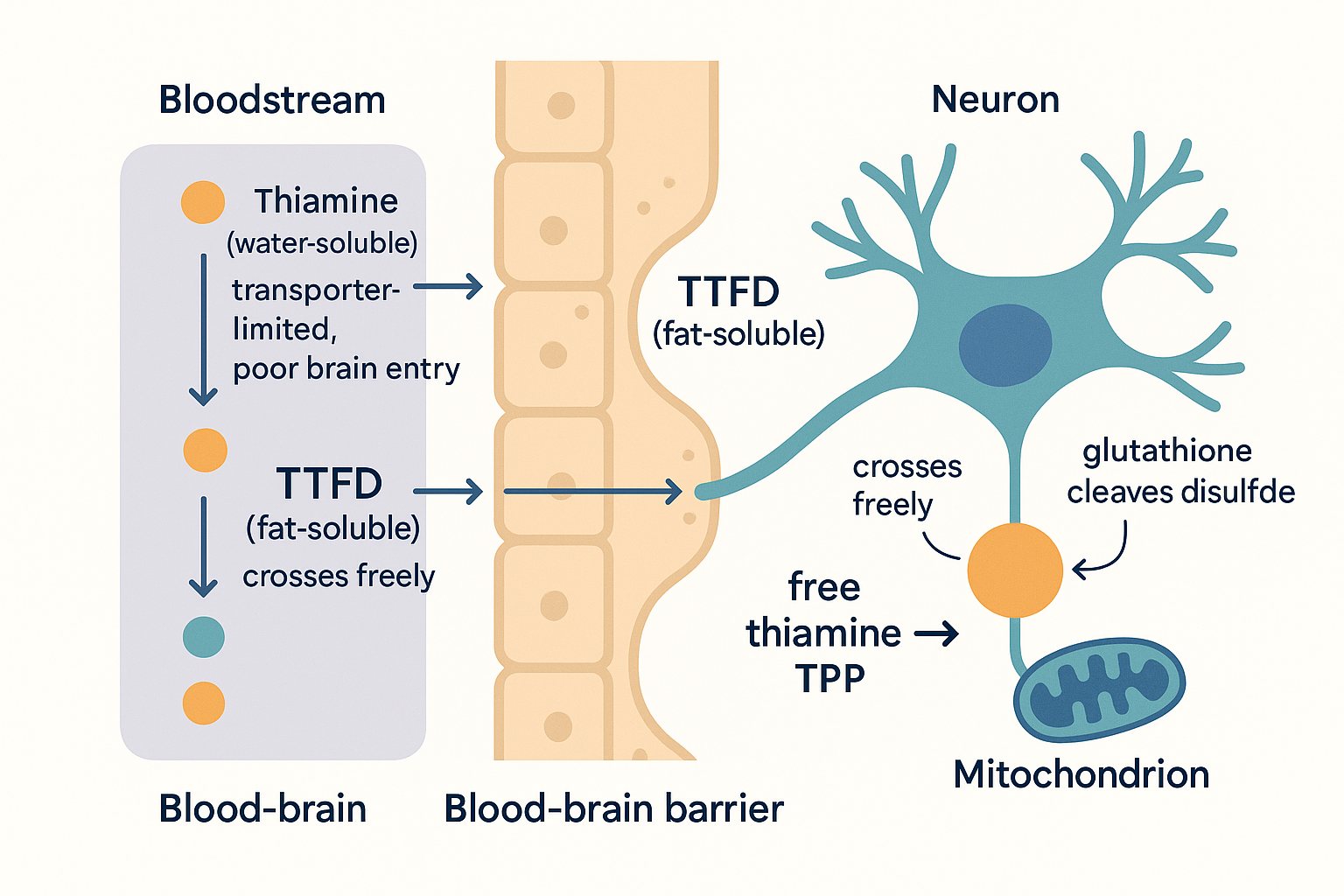 Why TTFD is the "brain form": ordinary thiamine is transporter-limited at the barrier, while fat-soluble TTFD diffuses straight through and self-converts to active TPP inside the neuron.
Why TTFD is the "brain form": ordinary thiamine is transporter-limited at the barrier, while fat-soluble TTFD diffuses straight through and self-converts to active TPP inside the neuron.
The natural analogue is allithiamine, the form of thiamine found in garlic (allium = garlic, hence the name). Garlic converts alliin + thiamine → allithiamine when the clove is crushed. This is partly why garlic has neurological associations in traditional medicine.
TTFD's CNS activity is confirmed by direct measurement: a first-in-human PET study (PMID 33812058) using [¹¹C]-labelled TTFD demonstrated accumulation in brain, pituitary, liver, kidney, and heart — with brain uptake superior to ordinary thiamine salts. Animal work (PMID 29992990) showed TTFD increases dopamine release in the medial prefrontal cortex via D1 receptor signalling, explaining some of the motivational and clarity effects users report beyond simple energy. Pharmacokinetic comparison (PMID 27707509) confirmed TTFD produces higher erythrocyte TDP concentrations than benfotiamine on an equivalent-mg basis.
Thiamax specific notes: - Standard dose: 25–50 mg/day (active), sometimes 100 mg for depletion - Start low: TTFD mobilises heavy metals (especially mercury) via the disulfide mechanism. This can cause paradox reactions — transient worsening of symptoms — in people with significant body burden. Elliot Overton has extensive documentation of this (search "TTFD paradox reaction EONutrition"). - Needs magnesium as cofactor — PDH, α-KGDH, and transketolase all require Mg²⁺. If magnesium is insufficient, more thiamine doesn't fully help. - Glutathione status matters — the intracellular conversion mechanism depends on it.
Sulbutiamine — the stimulant disulfide (not recommended for regular use)
Sulbutiamine is another disulfide thiamine (isobutyryl thiamine disulfide), also CNS-penetrant. It has a stimulant character — it increases dopaminergic and glutamatergic transmission in ways the other forms don't, and was a French pharmaceutical (Arcalion) used for asthenia.
The problem: chronic use downregulates THTR1, the thiamine transporter. It can worsen thiamine status with sustained use. It's more of an acute nootropic than a thiamine restoration tool.
Forms comparison
| Form | Fat-soluble | BBB crossing | Best for | Relative CNS effect |
|---|---|---|---|---|
| Thiamine HCl | No | Poor (transporter-limited) | Deficiency prevention only | Low |
| Thiamine mononitrate | No | Poor | Food fortification only | Low |
| Benfotiamine | Yes | Poor (converted before BBB) | Peripheral neuropathy, anti-glycation, metabolic | Low–medium |
| TTFD (Thiamax) | Yes | Excellent (passive diffusion into neurons) | CNS, cognitive, speech, energy | High |
| Allithiamine | Yes | Excellent | Same as TTFD | High |
| Sulbutiamine | Yes | Excellent | Acute fatigue (not chronic) | High (stimulant-type) |
Why your stack makes sense
High-carb morning → PDH bottleneck → thiamine clears it. Glucose floods into pyruvate, PDH needs more TPP to push it through, TTFD delivers TPP directly into mitochondria. More pyruvate becomes Acetyl-CoA instead of lactate. More Krebs cycle turns. More ATP. This is not placebo — the mechanism is linear.
Speech improvement → cerebellar + brainstem thiamine delivery. TTFD crosses the BBB and gets into cerebellar neurons. The timing circuits that sequence speech articulation have adequate TPP for α-KGDH. Glutamate excitotoxicity is modulated. Acetylcholine synthesis via Acetyl-CoA is less rate-limited. The output: slightly more precise and fluent speech, especially when the prior state was sub-optimal.
The reason this works better on high-carb/low-fat meals is also explicable: fat-dominant states (low-carb, ketosis) rely more on β-oxidation, which has lower thiamine demand per ATP produced. The bottleneck is felt most when you're running carbohydrate-heavy.
Cofactors — thiamine doesn't work alone
PDH and α-KGDH are multi-enzyme complexes that require multiple cofactors beyond TPP:
| Cofactor | Why it matters |
|---|---|
| Magnesium (Mg²⁺) | Required by all three TPP-dependent enzymes. Deficiency mimics thiamine deficiency. |
| Lipoic acid | Component of PDH and α-KGDH complexes (E2 subunit) |
| Riboflavin (B2) | FAD-dependent subunit of both complexes (E3) |
| Niacin (B3) | NAD⁺-dependent step (E3 again) |
| Glutathione | Required for intracellular TTFD conversion to free thiamine |
If you're getting excellent results from TTFD alone, you probably already have adequate magnesium and B2. But if effects plateau, adding magnesium glycinate or malate (200–400 mg elemental Mg/day) and riboflavin (10–50 mg/day) often extends the benefit.
Who needs more thiamine than the RDA
The RDA (1.1–1.2 mg/day) is calculated for minimal deficiency prevention. It's not calibrated for high-carb diets, heavy exercise, chronic stress, or neurological optimisation.
Higher demand in: - High-carbohydrate diets (more pyruvate load through PDH) - Heavy alcohol use (alcohol interferes with thiamine absorption and storage directly — Wernicke's is an alcoholism-related syndrome) - High-intensity exercise (increased mitochondrial turnover) - Chronic stress (adrenal function burns B vitamins) - Raw fish or shellfish consumption (thiaminase enzyme in raw flesh destroys thiamine) - Genetic THTR2 variants (SLC19A3 polymorphisms reduce active absorption) - Proton pump inhibitor use (changes gut pH, affects absorption)
Practical approach
Goal: cognitive / energy / speech (your use case) → TTFD (Thiamax) 25–50 mg with your high-carb morning meal → Add magnesium glycinate 200–400 mg if not already taking → Start at 25 mg; if paradox reaction (worsening, detox-like symptoms), drop to 12.5 mg and build up
Goal: peripheral neuropathy / blood sugar / anti-glycation → Benfotiamine 150–300 mg/day → Can combine with TTFD for both peripheral and central coverage
Goal: deficiency prevention only → Any form at low dose works. Benfotiamine is more cost-effective than TTFD for this purpose.
Avoid thiamine HCl at high doses — the dose-response curve flattens above ~10 mg due to transporter saturation. If you're taking 100 mg+ of thiamine HCl, you're largely paying to pee it out.
UK sourcing
| Product | Form | Notes |
|---|---|---|
| Thiamax (Objective Nutrients) | TTFD 25 mg | Elliot Overton's formulation, ships to UK, most clinically studied TTFD product |
| Benfotiamine (Bulk, Solgar, Source Naturals) | Benfotiamine 150–300 mg | Widely available on Amazon UK |
| Now Foods Benfotiamine | Benfotiamine 150 mg | Budget-friendly, consistent |
| Allithiamine (Ecological Formulas) | Natural allithiamine | Available via Evitamins/iHerb, natural TTFD analogue |
Research sources
TTFD / CNS penetration - PMID 33812058 — First-in-human PET study with [¹¹C]-TTFD. Direct imaging: TTFD accumulates in brain, pituitary, liver. Brain uptake superior to water-soluble thiamine salts. Biochem Biophys Res Commun (2021). - PMID 29992990 — Saiki et al. TTFD increases dopamine release in medial prefrontal cortex via D1 receptor signalling; effect blocked by D1 antagonist. Scientific Reports (2018). - PMID 27707509 — Park et al. Fursultiamine (TTFD) vs benfotiamine pharmacokinetics; TTFD produced higher erythrocyte TDP concentrations. Clinical Therapeutics (2016). - PMID 12195231 — Lonsdale, Shamberger, Audhya. Pilot: 8/10 autistic children improved on TTFD suppositories 50 mg bid × 2 months. Neuro Endocrinol Lett (2002).
Benfotiamine bioavailability - PMID 24399744 — Xie et al. Plasma thiamine AUC after benfotiamine: ~1147% vs thiamine HCl; erythrocyte TDP ~196% vs HCl. J Clin Pharmacol (2014). - PMID 8929745 — Loew D. Peak plasma ~5×, bioavailability ~3.6× vs thiamine HCl; transketolase activity increased only after benfotiamine. Int J Clin Pharmacol Ther (1996). - PMID 9587048 — Greb & Bitsch. 7-volunteer crossover: benfotiamine superior in plasma and haemolysate; thiamine disulfide had lowest bioavailability of all forms. Int J Clin Pharmacol Ther (1998). - PMID 9638312 — Hilbig & Rahmann. Autoradiography in mice: benfotiamine showed 5–25× higher brain tracer incorporation vs thiamine HCl. (Note: vs HCl only, not vs TTFD.) Arzneimittelforschung (1998).
Thiamine transporters (THTR1/THTR2) - PMID 23506878 — SLC19 transporter family review. THTR2 (SLC19A3) Km ~25 nM (very high affinity); THTR1 (SLC19A2) Km ~2.5 µM. PMC (2013). - PMID 11731220 — Rajgopal et al. SLC19A3 characterisation: saturable, selective, pH-dependent thiamine transport. Biochim Biophys Acta (2001). - PMID 22305197 — Smithline, Donnino, Greenblatt. Pharmacokinetics of 100/500/1500 mg oral thiamine HCl in healthy volunteers: plasma Cmax continued rising at all doses; absorption is not fully saturated but efficiency per mg falls sharply above the active-transport range. BMC Clin Pharmacol (2012). - PMID 6464934 — Saturable kinetics dominate below ~1.5 µM luminal thiamine; linear passive diffusion takes over above ~2 µM. J Nutr (1984).
Alpha-KGDH and neurotransmitters - PMID 36293260 — Hansen & Gibson. KGDHC as hub of neural plasticity: TPP-dependence, glutamate/GABA dysregulation, selective neurodegeneration. Int J Mol Sci (2022). - PMID 17482317 — Shi et al. KGDHC activity fell 52% in submedial thalamic nucleus (vulnerable region) in thiamine-deficient rats. Neurochem Int (2007). - PMID 6149477 — Gibson et al. K⁺-stimulated ACh synthesis fell ~75%; KGDHC more sensitive than PDH in brain. Fully reversed by 7-day thiamine repletion. Neurochem Res (1984).
PDH - PMID 3139833 — Elnageh & Gaitonde. PDH holoenzyme protein preserved in thiamine-deficient rat brain; limitation is TPP cofactor availability, not enzyme degradation. J Neurochem (1988).
Cerebellar vulnerability / Wernicke's - PMID 11677067 — Murata et al. Serial MRI/MRS of Wernicke's: thalamic lesions reversed by thiamine; cerebellar lesions may be permanent. Psychiatry Research (2001). - PMID 37048595 — Arts et al. Post-COVID Wernicke's: severe cerebellar ataxia (anterior vermal atrophy) without dysarthria or upper limb ataxia. J Clin Med (2023).
Non-peer-reviewed - Elliot Overton (EONutrition / eonutrition.co.uk) — Extensive clinical case documentation of TTFD dosing, paradox reactions, heavy metal mobilisation, and dysautonomia protocols. Practitioner-level; synthesises Lonsdale's peer-reviewed TTFD work with clinical observations. Not confirmatory evidence but the primary English-language practical resource on TTFD.