Aspirin
One of the oldest drugs in the world, a few cents a dose — and far stranger than its "painkiller" label suggests. How aspirin actually works at the molecular level: the irreversible enzyme acetylation behind its anti-inflammatory, anti-clotting, and anti-fever effects, and the genuinely real (and genuinely double-edged) metabolic effects that the Ray Peat world prizes it for.
Read alongside
Aspirin's main action is inhibiting the COX → prostaglandin arm of inflammation, so the Inflammation foundations page is the key companion. Its metabolic effects lean on the uncoupling and AMPK biology of Cellular Energy, and it appears as an anti-aromatase tool in the Hormones page and the Testosterone Kabbalah.
Why aspirin is more interesting than it looks
Aspirin is so familiar — the headache pill, the heart-attack tablet — that it's easy to assume it's fully understood and boring. It is neither. The biohacking commentary (e.g. @BasedBiohacker, drawing on decades of Ray Peat's writing) makes a provocative claim: aspirin "is not a pain drug, it's a metabolic drug." That overstates it — aspirin is genuinely a pain and anti-clotting drug — but it points at something real: aspirin has effects on mitochondrial metabolism, the energy-sensing system, fat metabolism, and oestrogen that go well beyond "blocks pain," and that are mechanistically traceable.
This deep dive separates the rock-solid pharmacology (the COX mechanism, taught in every medical school) from the more speculative bioenergetic claims (the "pro-thermogenic, thyroid-mimetic" framing) — explaining how each works, and flagging where enthusiasm outruns evidence. Because aspirin is also a real drug with real dangers, the risks get their own honest section.
What aspirin actually is
Aspirin is acetylsalicylic acid. Its story starts with salicylate — a compound in willow bark, used for pain and fever since antiquity (Hippocrates mentioned willow). The problem with raw salicylic acid was that it savaged the stomach. In 1897, a chemist at Bayer (Felix Hoffmann) added an acetyl group to salicylic acid, producing acetylsalicylic acid — gentler on the gut and more effective. That acetyl group, added for tolerability, turns out to be the key to aspirin's unique mechanism, as we'll see.
Once swallowed, aspirin is rapidly broken down in the body back into salicylate (its main active metabolite) plus the freed acetyl group. So aspirin really has two active identities: the intact acetylsalicylic acid (which does the special irreversible trick) and the salicylate it becomes (which does much of the metabolic work). Keep both in mind.
The core mechanism: irreversibly switching off COX
Aspirin's central, Nobel-Prize-winning mechanism (John Vane, 1982) is the inhibition of an enzyme called cyclooxygenase (COX). This single action explains most of its classic effects, so it's worth understanding precisely.
Recall from the Inflammation page the eicosanoid arm of inflammation: a polyunsaturated fat called arachidonic acid, released from cell membranes, is converted by COX into prostaglandins and thromboxanes — local signalling molecules that drive pain, fever, inflammation, and blood clotting. COX is the gateway enzyme of that whole pathway.
Aspirin blocks COX — and it does so in a way no other drug does. While ibuprofen and other NSAIDs sit in the enzyme's active site reversibly (they come and go), aspirin uses its acetyl group to chemically bolt itself onto the enzyme — it acetylates a specific amino acid (serine 530) in COX's active site. This is a permanent, irreversible modification: that COX molecule is dead for good. The enzyme can only be replaced by the cell synthesising a brand-new one. This irreversibility is the source of aspirin's distinctive properties.
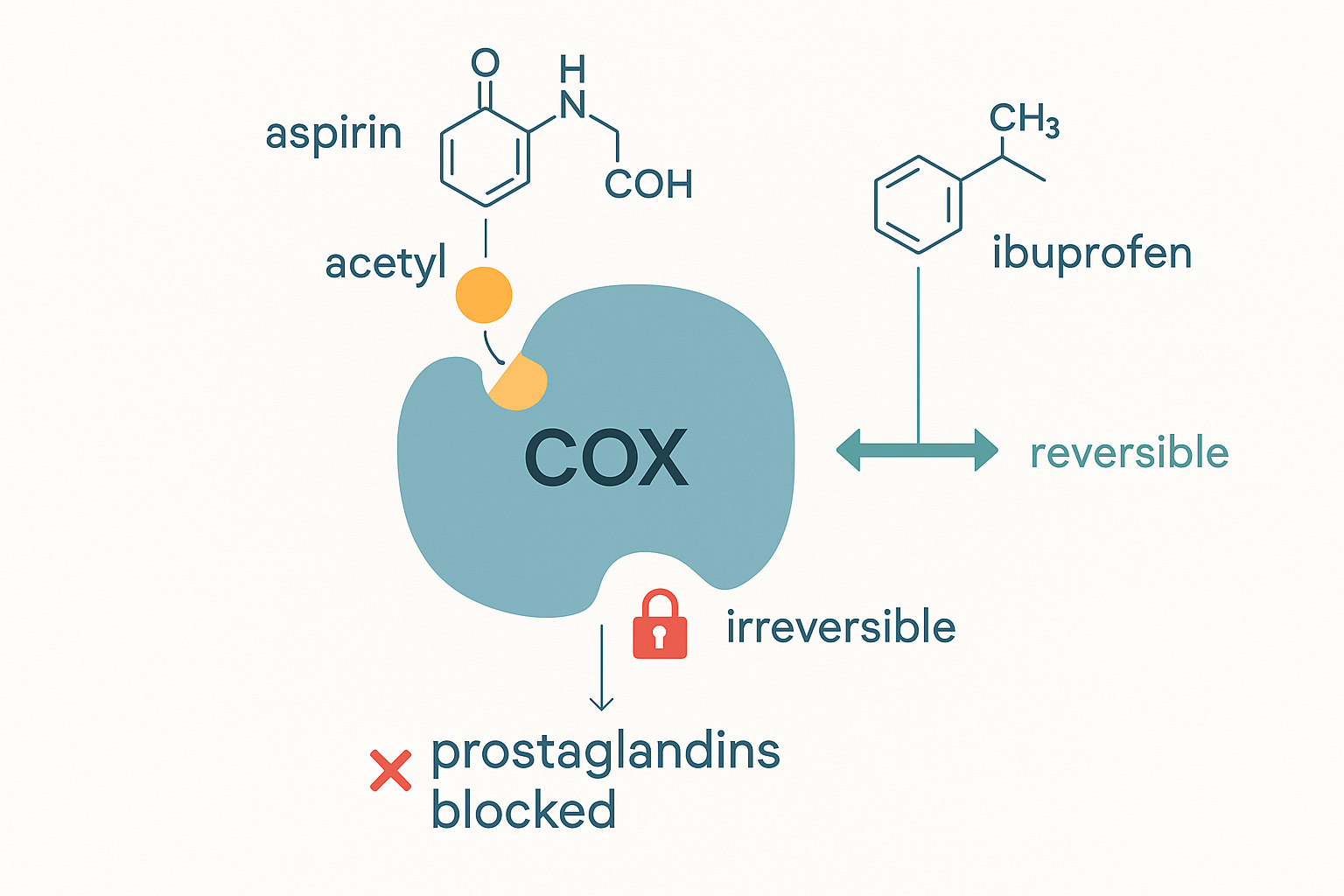 Aspirin's unique trick: it bolts its acetyl group permanently onto serine-530 in the COX active site — unlike reversible NSAIDs that come and go — killing that enzyme for good and shutting down the prostaglandin/thromboxane pathway.
Aspirin's unique trick: it bolts its acetyl group permanently onto serine-530 in the COX active site — unlike reversible NSAIDs that come and go — killing that enzyme for good and shutting down the prostaglandin/thromboxane pathway.
There are two forms of the enzyme, and the distinction matters for every effect below:
- COX-1 — the "housekeeping" form, present all the time, making prostaglandins for protective everyday jobs: maintaining the stomach lining, kidney blood flow, and platelet function.
- COX-2 — the "inducible" form, switched on by injury and inflammation, making the prostaglandins of pain, fever, and swelling (and, relevantly, driving aromatase — see below).
flowchart TD
MEM["Cell membrane PUFA<br/>(arachidonic acid)"] --> COX{COX enzyme}
ASA["Aspirin<br/>(irreversibly acetylates COX)"] -. blocks .-> COX
COX -->|COX-1 housekeeping| PG1["Protective prostaglandins:<br/>stomach lining · kidney · platelets"]
COX -->|COX-2 inducible| PG2["Inflammatory prostaglandins:<br/>pain · fever · swelling · aromatase"]
COX --> TXA["Thromboxane A2<br/>(platelet clotting)"]From this one mechanism, every classic effect falls out:
- Anti-inflammatory & anti-pain — blocking COX-2 cuts the inflammatory prostaglandins that sensitise nerves and drive swelling. This is the direct, evidence-solid sense in which aspirin "reduces inflammation": it amputates one arm of the inflammation cascade.
- Anti-fever (antipyretic) — fever is set by prostaglandins acting on the hypothalamus (the inflammation page's sickness-behaviour mechanism); block their production and the fever set-point drops.
- Anti-clotting (antiplatelet) — this is where irreversibility shines. Platelets (which trigger clots) make thromboxane A2 via COX-1 to clump together. Aspirin permanently kills COX-1 in platelets — and platelets have no nucleus, so they cannot make new COX. A single low dose disables a platelet's clotting machinery for its entire ~10-day lifespan. This is why a tiny daily "baby aspirin" (75–100 mg) thins the blood and prevents heart attacks and strokes: it's exploiting the platelet's inability to recover.
The metabolic angle: where it gets interesting (and contested)
Now the claims the bioenergetic world cares about — that aspirin "stimulates mitochondrial respiration," is "pro-thermogenic," and "mirrors thyroid hormone." These are not fabrications, but they are the salicylate (not the COX) side of the story, and they are genuinely double-edged. Here is the real biochemistry behind each:
1. Salicylate is a mild mitochondrial uncoupler. Recall uncoupling from the cellular energy page: if the mitochondrial proton gradient is allowed to leak back without driving ATP synthase, the energy is released as heat, and the electron transport chain runs faster to compensate — consuming more oxygen. Salicylate does exactly this — it mildly uncouples oxidative phosphorylation. This is the real mechanism behind "pro-thermogenic" and "increases mitochondrial oxygen consumption": at modest doses it nudges the cell toward burning more fuel as heat. Crucially, this is the same mechanism that makes salicylate overdose dangerous — aspirin poisoning causes hyperthermia (dangerous overheating) and metabolic acidosis precisely because the uncoupling runs out of control. So "pro-thermogenic" is true and double-edged: a feature at low dose, a toxicity at high dose.
2. Salicylate directly activates AMPK. AMPK (from the cellular energy page) is the master energy-sensor — it's switched on when the cell needs to make more energy and burn fuel, and it's the same enzyme activated by exercise and by the diabetes drug metformin. A 2012 study (Science, Hawley et al.) showed salicylate directly binds and activates AMPK — a clean molecular basis for aspirin's metabolic, fat-burning, and insulin-sensitising effects. This is solid science, not Peat folklore.
3. It lowers free fatty acids (anti-lipolytic). Salicylate inhibits lipolysis — the release of stored fat into the blood as free fatty acids (FFAs). The bioenergetic relevance (and the BasedBiohacker "puffiness/water retention" point) connects to two earlier threads: lower circulating FFAs means less of the polyunsaturated fat that gets peroxidised (Vitamin E and inflammation pages), and via the Randle cycle (cellular energy), fewer FFAs competing means better glucose oxidation — the "cleaner-burning" state the Peat framework prizes.
4. "Mirrors thyroid hormone." This is an interpretation, not a distinct mechanism: because uncoupling + AMPK + better glucose oxidation all raise metabolic rate and body heat, the net effect resembles what thyroid hormone (T3) does (the metabolism page). It's a reasonable analogy for the phenotype, but aspirin does not act on thyroid receptors — it reaches a similar destination by a different road.
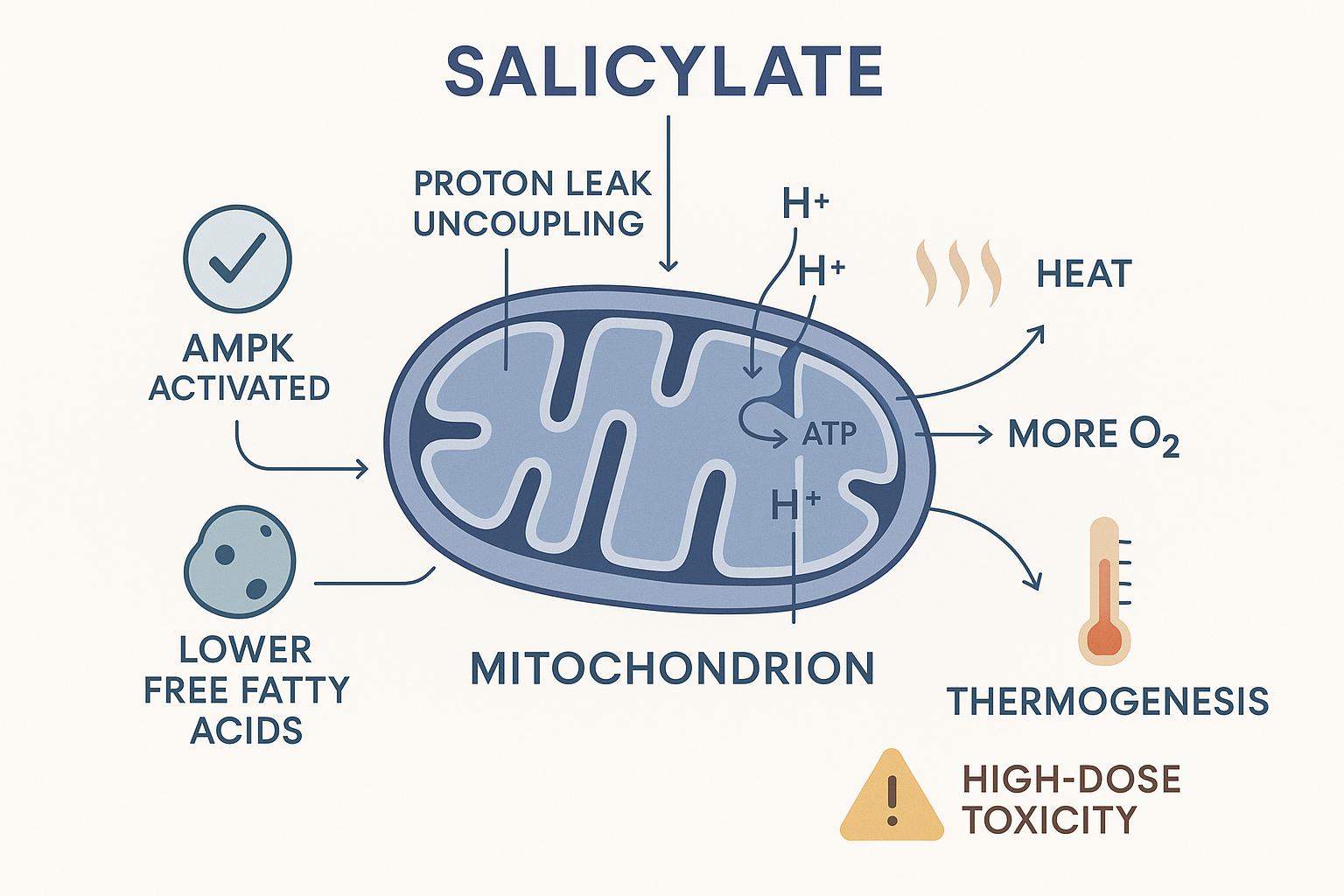 The salicylate side: mild mitochondrial uncoupling (heat, more oxygen burned), AMPK activation, and lower free fatty acids together nudge up metabolic rate — the kernel of truth behind "mirrors thyroid." The very same uncoupling is what makes overdose dangerous.
The salicylate side: mild mitochondrial uncoupling (heat, more oxygen burned), AMPK activation, and lower free fatty acids together nudge up metabolic rate — the kernel of truth behind "mirrors thyroid." The very same uncoupling is what makes overdose dangerous.
⚖️ Calibration. The COX mechanism is textbook-solid. The metabolic claims are real but smaller and more two-sided than the thread implies: the uncoupling that makes aspirin "thermogenic" is the very thing that makes overdose deadly; the AMPK activation is genuine but its whole-body significance at normal doses is modest; the FFA-lowering is real. "Aspirin is a metabolic drug that mirrors thyroid" is a stylish overstatement of a kernel of truth. It nudges metabolism; it does not replace a thyroid.
Anti-oestrogen and anti-cancer effects
Two more effects, both flowing largely from COX-2 inhibition, both with real evidence:
- Anti-aromatase / anti-oestrogen. COX-2 drives the expression of aromatase — the enzyme (from the Hormones page) that converts testosterone to oestrogen. By inhibiting COX-2, aspirin lowers aromatase activity and oestrogen production. This is exactly why aspirin appears on the Testosterone Kabbalah's list of oestrogen-lowering tools, and it's a documented mechanism (much of the oestrogen-lowering benefit of aspirin in breast-cancer epidemiology runs through this).
- Cancer chemoprevention. Long-term aspirin use is associated with reduced risk of colorectal (and some other) cancers — among the best-supported "off-label" benefits of any drug. The mechanism is again COX-2/prostaglandin: tumour-promoting inflammation runs partly through COX-2, and aspirin's irreversible block (plus AMPK activation) is anti-proliferative. This is mainstream, not fringe.
The Peat protocol and the stack (with the reasons)
The BasedBiohacker thread relays a fairly standard bioenergetic-aspirin protocol. The practical points, with the why attached:
- Dose: ~325 mg (one regular aspirin) daily is a common starting point; some use lower (100–200 mg). Peat himself reportedly used it regularly but not high-dose chronically — sensible, given the uncoupling-toxicity at high dose.
- Always with food, never empty stomach — because COX-1 inhibition removes the protective stomach prostaglandins, the leading risk is gastric irritation and ulcers (see risks). Food and lower doses mitigate this.
- Timing: bedtime dosing for overnight inflammation/sleep; morning for daytime metabolic/energy effects.
- Light cycling (a day or two off per week) for long-term use — not strictly required, but reasonable.
And the stack — aspirin is explicitly not taken alone, and each partner has a real rationale:
- Vitamin K2 — the most important one. Aspirin thins the blood and antagonises vitamin K-dependent clotting; K2 also keeps calcium in bones and out of arteries. Pairing addresses both the clotting and the calcium-handling concerns. (This is a genuine safety-relevant pairing, not just optimisation.)
- Magnesium — supports the mitochondrial/metabolic effects (every ATP step, per the magnesium deep dive) and is somewhat GI-protective.
- Glycine / gelatin — protective for the gut lining and connective tissue that aspirin can irritate; anti-inflammatory in its own right.
- Vitamin C with bioflavonoids — antioxidant support; Peat often paired them, and vitamin C protects the gut and regenerates other antioxidants (Vitamin E deep dive).
- Caffeine / black coffee — taken together in the classic Peat "aspirin + coffee" combination, both pushing toward the higher-metabolic, pro-thyroid state.
The risks — honestly
Aspirin is cheap and old, but it is a real drug with real, sometimes serious harms. This is non-negotiable context:
- Gastrointestinal bleeding and ulcers — the big one. By killing COX-1, aspirin removes the prostaglandins that protect the stomach lining, so chronic use causes erosions, ulcers, and bleeding (sometimes silent and serious). Risk rises with dose, age, alcohol, and on an empty stomach.
- General bleeding risk — the same antiplatelet effect that prevents clots makes bleeding (including brain bleeds) more likely and harder to stop. This is why daily aspirin for primary prevention has fallen out of favour for many people — the bleeding risk can outweigh the clot-prevention benefit.
- Reye's syndrome — aspirin must not be given to children/teenagers with viral illness (flu, chickenpox); it can trigger this rare, often-fatal swelling of the liver and brain. (Aspirin is an adult drug.)
- Salicylate toxicity — at high doses, the very uncoupling that's "thermogenic" becomes dangerous: tinnitus (ringing ears) is the early warning, escalating to hyperthermia, metabolic acidosis, and worse. The dose-response is real.
⚖️ Calibration — bottom line. Aspirin is a genuinely remarkable molecule: a well-understood, irreversible COX inhibitor (rock-solid for inflammation, pain, fever, and clot prevention) with real, mechanistically-traceable metabolic effects (mild uncoupling, AMPK activation, lower FFAs, lower oestrogen) that justify the bioenergetic interest. But the "metabolic drug, not a pain drug" framing oversells a kernel of truth, and the same mechanisms that make it "thermogenic" make overdose dangerous. It is not a casual everyday supplement — the GI-bleeding and bleeding risks are real, the K2 pairing matters, and "with food, modest dose, not high-dose chronic" is the defensible approach. As always: understand the mechanism, respect the drug, and this is not medical advice.
Related compounds & foundations
The aspirin stack
- Aspirin — the compound page.
- Vitamin K2 — the key safety pairing (clotting + calcium handling).
- Magnesium — supports the metabolic effects; see the magnesium deep dive.
- Glycine / gelatin — gut and connective-tissue protection.
- Vitamin C, caffeine — the classic Peat antioxidant and pro-metabolic pairings.
Mechanistically related
- Vitamin E — the other major anti-inflammatory/anti-peroxidation tool; aspirin lowers the FFAs that peroxidise.
- Niacinamide — another anti-lipolytic, pro-metabolic Peat staple.
Related foundations
- Inflammation — the COX → prostaglandin arm aspirin blocks.
- Cellular Energy — uncoupling and AMPK, the basis of aspirin's metabolic effects.
- Hormones & the Endocrine System — aromatase, the enzyme aspirin lowers via COX-2.
- Systemic Metabolism — the thyroid/thermogenesis phenotype aspirin partly mimics.
- The Testosterone Kabbalah — where aspirin features as an oestrogen-lowering lever.
Source: thread by @BasedBiohacker (Ray Peat-influenced aspirin protocol). The COX/acetylation pharmacology, AMPK activation (Hawley et al., Science 2012), uncoupling, and anti-aromatase mechanisms reflect established research; the "metabolic drug / mirrors thyroid" framing is flagged as a stylised interpretation, and the risks section reflects standard clinical pharmacology.